AutoDock Vina Protein-Ligand Docking Pipeline
Project Overview
This portfolio project demonstrates a containerized pipeline for computational drug discovery using molecular docking. The workflow is implemented as a collection of interconnected Docker containers (metaframes) that together form a complete docking workflow. Each metaframe performs a specific task in the molecular docking pipeline, communicating with other metaframes through a standardized interface. The system includes both processing and visualization components, creating a comprehensive and interactive drug discovery platform.
Technical Implementation
Architecture
The pipeline uses a microservices architecture with Docker containers as the primary deployment mechanism. Each service (metaframe) is designed to perform a specific task in the docking workflow and communicates through a standardized I/O interface. This approach offers several advantages:
- Modularity: Individual components can be updated or replaced without affecting the entire pipeline
- Scalability: Computationally intensive steps can be scaled independently
- Technology flexibility: Different tools and languages can be used for different steps
- Reproducibility: Docker ensures consistent environments
- Interactive visualization: Dedicated visualization metaframes provide real-time feedback
Technology Stack
- JavaScript: Used for PDB file retrieval, 3D visualization (with NGL Viewer), and interactive results display
- Python: Used for data processing, ChEMBL API interaction, and results analysis
- Bash: Used for workflow orchestration and gluing components together
- Docker: Container platform for deployment
- OpenBabel: Chemical file format conversion
- AutoDock Vina: Molecular docking engine
- RDKit: Cheminformatics toolkit for molecule manipulation
- Meeko: Ligand preparation for docking
- NGL Viewer: 3D molecular visualization
Key Metaframes
Processing Metaframes
PDB File Retrieval (JavaScript)
- Fetches protein structure data from the RCSB PDB database
- Implemented as a simple JavaScript application using the Fetch API
- Handles error conditions gracefully
Protein-Ligand Separation (Bash + OpenBabel)
- Processes PDB files to separate protein and ligand components
- Uses grep pattern matching to extract relevant atom records
- Converts ligand to SDF format using OpenBabel
Compound Library Generation (Python + OpenBabel)
- Converts ligand to SMILES format
- Queries the ChEMBL API for similar compounds
- Transforms 2D structures to 3D and prepares them for docking
- Implements error handling and fallback mechanisms
Structure Preparation (Python + OpenBabel + Meeko)
- Prepares protein and ligand structures for molecular docking
- Adds hydrogens and computes partial charges
- Generates PDBQT files required by AutoDock Vina
- Implements multiple preparation methods with fallbacks
Molecular Docking (AutoDock Vina)
- Calculates binding box parameters from reference ligand
- Splits compound library into individual ligands
- Performs docking calculations for each ligand
- Outputs binding poses and scores
Results Analysis (Python)
- Extracts binding affinities from docking logs
- Generates a sortable CSV report
- Creates an interactive HTML visualization
- Implements score normalization for visual comparison
Complex Generation (Python + OpenBabel)
- Creates 3D structural models of protein-ligand complexes
- Converts AutoDock output to standard PDB format
- Generates visualization-ready files
- Optimizes geometry for proper display
Visualization Metaframes
Raw PDB Viewer
- Displays the raw structure file contents
- Allows examination of atomic coordinates
- Provides basic file inspection capabilities
Protein-Ligand Visualizer
- Shows the original protein with its co-crystallized ligand
- Provides 3D interactive view of starting structures
- Helps understand the binding site context
Protein Structure Visualizer
- Dedicated view of protein structure alone
- Highlights secondary structure elements
- Allows different rendering styles (cartoon, surface, etc.)
Ligand Structure Visualizer
- Shows reference ligand structure in 3D
- Provides atom-level visualization
- Assists in understanding target binding interactions
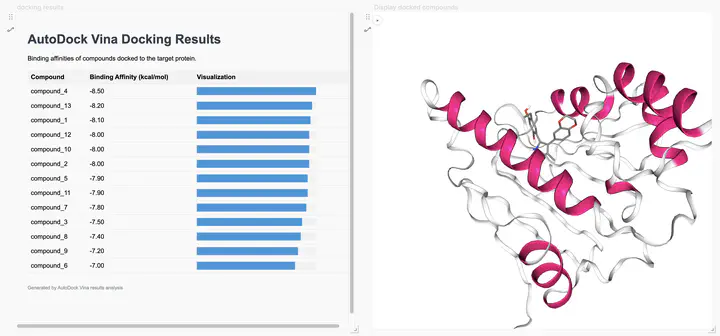
Docking Results Visualizer
- Displays sortable table of docking scores
- Shows interactive binding energy comparisons
- Presents ranking of compounds by predicted affinity
- Implements a generic HTML renderer for flexible report display
- Handles both direct HTML content and URL-based HTML references
Docked Compounds Visualizer
- Interactive 3D view of docked compounds in protein binding site
- Allows visual inspection of binding poses
- Helps identify key protein-ligand interactions
- Supports comparative visualization of multiple compounds
Scientific Background
Virtual Screening and Molecular Docking
Molecular docking is a computational technique used in drug discovery to predict the binding orientation and affinity of small molecules (ligands) to protein targets. This pipeline implements a structure-based virtual screening workflow, which follows these general steps:
- Target preparation: A protein structure is obtained and prepared for docking
- Ligand preparation: Chemical compounds are processed to generate 3D coordinates and proper protonation states
- Docking: Each compound is docked to the protein target to generate binding poses
- Scoring: Binding poses are evaluated and ranked based on predicted binding energy
- Analysis: Results are analyzed to identify promising compounds for further study
- Visualization: Binding poses are visualized to understand protein-ligand interactions
AutoDock Vina
AutoDock Vina is a widely-used open-source molecular docking program. It offers:
- Accurate binding predictions
- Fast performance compared to earlier alternatives
- Support for flexible ligands and (limited) receptor flexibility
- Multithreading capabilities
- A scoring function based on empirical free energy calculations
Challenges and Solutions
Challenge 1: File Format Compatibility
Problem: Different tools in the pipeline require different molecular file formats.
Solution: Implemented conversion utilities using OpenBabel, with careful attention to maintaining chemical information (atom types, bond orders, etc.) through each conversion.
Challenge 2: Reliability of External APIs
Problem: The ChEMBL API can sometimes be unreliable or return unexpected data structures.
Solution: Implemented robust error handling with multiple fallback mechanisms:
- Multiple methods to extract SMILES data from responses
- Detailed logging for troubleshooting
- Alternative approaches when primary methods fail
Challenge 3: Docking Box Determination
Problem: AutoDock Vina requires specification of a search space (docking box), which significantly impacts results.
Solution: Created an automated approach to calculate optimal docking box parameters:
- Uses reference ligand to determine binding site location
- Adds appropriate buffer space around the ligand
- Falls back to reasonable defaults when reference information is unavailable
Challenge 4: Processing Large Compound Libraries
Problem: Docking large compound libraries can be time-consuming and error-prone.
Solution: Implemented a batch processing approach:
- Splits compound libraries into individual files
- Processes each compound independently
- Consolidates results after docking
- Provides comprehensive logging and error reporting
Challenge 5: Interactive Visualization
Problem: Effective visualization of 3D molecular structures requires specialized tools and optimized data formats.
Solution: Developed a multi-layer visualization approach:
- Used NGL Viewer for interactive 3D visualization
- Created proper protein-ligand complexes for visualization
- Implemented comparative visualization tools
- Ensured consistent coloring and representation schemes
Challenge 6: Workflow Integration
Problem: Coordinating data flow between independent, containerized components.
Solution: Designed a standardized I/O interface:
- Used well-defined file formats for data exchange
- Implemented consistent naming conventions
- Created robust error handling for inter-metaframe communication
- Designed fail-safe mechanisms when metaframes received unexpected inputs
Future Enhancements
- Parallelization: Implement multi-node processing for large compound libraries
- Machine Learning Integration: Add ML-based scoring functions to re-rank compounds
- Molecular Dynamics: Extend the pipeline to include MD simulations of top compounds
- Web Interface: Create a web-based front-end for easier visualization and interaction
- Results Database: Add a database backend to store and query docking results
- Interactive Reports: Enhance visualization with interactive 3D reports
- Binding Site Analysis: Add tools for analyzing protein binding site characteristics
- Integration with Public Databases: Connect to additional compound databases beyond ChEMBL
Conclusion
This project demonstrates a practical approach to computational drug discovery using modern containerization technology. By breaking the workflow into discrete, reusable components, it provides a flexible, maintainable, and scalable platform for virtual screening campaigns. The modular architecture allows for easy adaptation to different targets and compound libraries, making it a valuable tool for both research and educational purposes.
The implementation showcases expertise in:
- Scientific software development
- Containerization and microservices
- API integration
- Error handling and robust system design
- Chemical informatics
- Computational drug discovery methods
- Interactive data visualization
- Complex workflow orchestration
Karl Lundquist
Bioinformatics Data Scientist
I am passionate about leveraging artificial intelligence and machine learning to tackle complex problems related to human health.